Revista Paraguaya de Reumatología, Vol. 2, N° 1, 2016
ISSN 2413-4341
CASO CLÍNICO
ENFERMEDAD RELACIONADA A IGG4. REPORTE DEL PRIMER CASO REGISTRADO EN PARAGUAY
Juan Gabriel Elizaur López, Yanira Rosana Yinde, Julio Daniel Mazzoleni Insfrán
Hospital Central Dr. Emilio Cubas, Instituto de Previsión Social, Asunción, Paraguay
Servicio de Reumatología, Departamento de Medicina Interna, Hospital Central del Instituto de Previsión Social, Asunción, Paraguay
Autor para correspondencia. Correo electrónico: gabrielelizaur13@hotmail.com (J. G. Elizaur López)
RESUMEN
La enfermedad relacionada a IgG4 es una entidad sistémica, inmunomediada, de características fibroinflamatorias y de etiología desconocida, capaz de afectar cualquier órgano de la economía, hasta su insuficiencia funcional. Los órganos frecuentemente afectados son: páncreas, vías biliares, mamas, las glándulas salivales y lacrimales, entre otros. Las manifestaciones clínicas dependen del órgano afectado. La incidencia exacta a nivel mundial es desconocida, aunque se estima que es baja. El diagnóstico se sospecha fundamentalmente por aspectos clínico-radiológicos y se confirma por la presencia de patrones histológicos típicos tras haber descartado otras patologías más comunes. El tratamiento está dirigido a disminuir o evitar la progresión de fibrosis con el empleo de corticoides y/o agentes inmunosupresores. Se presenta el primer caso registrado de enfermedad relacionada a IgG4 en Paraguay.
Palabras claves: Enfermedad relacionada a IgG4, pseudotumor inflamatorio, pancreatitis autoinmune
IGG4 RELATED DISEASE. CASE REPORT OF FIRST CASE REPORTED IN PARAGUAY
ABSTRACT
IgG4 related disease is a systemic, inmunomediated entity of fibroinflammatory features and unknown etiology, capable of targeting any organ of the economy promoting its functional insufficiency. Organs frequently affected are: pancreas, gallbladder and coleducus, breast, salivary and lacrimal glands, among others. Clinical manifestations depend upon the organ involved. The exact global incidence is unknown but is estimated as low. The diagnosis is suspected upon clinical and radiologic features, and confirmed through the presence of typical histopathological patterns after ruling out other more frequent pathologies. Treatment is aimed at reducing or inhibiting the progression of fibrosis with the use of corticosteroids and inmunosuppresants. We present here the first registered case of IgG4 related disease in Paraguay.
Keywords: IgG4-related disease, inflammatory pseudo-tumor, autoimmune pancreatitis
INTRODUCCIÓN
La enfermedad relacionada a la inmunoglulina G tipo cuatro (ER-IgG4) es una entidad sistémica, inmunomediada, de características fibroinflamatorias y de etiología desconocida, capaz de afectar cualquier órgano de la economía, hasta su insuficiencia funcional. Los órganos frecuentemente afectados son: páncreas, vías biliares, mamas, las glándulas salivales y lacrimales, entre otros1.
La incidencia a nivel internacional es baja, con predominio en varones caucásicos en proporción de 2,8:1 y las descripciones fueron en su mayoría en japoneses con pancreatitis autoinmune tipo I, que es el actual prototipo de la enfermedad2,3. Entre los diagnósticos diferenciales se pueden citar la tiroiditis de Riedel, la aortitis linfoplasmocítica, la fibrosis retroperitoneal idiopática, la enfermedad de Mickulicz y el demoninado tumor de Küttner, entre otros1,3–5.
CASO CLINÍCO
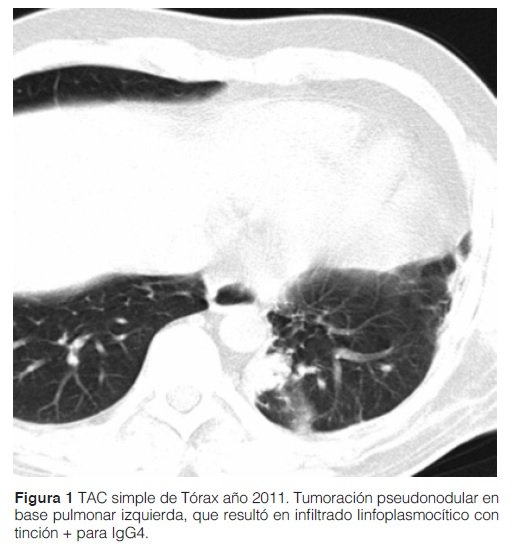
Paciente de sexo masculino de 32 años, sin patología de base, que consulta por pérdida de peso, fiebre de origen desconocido (FOD), que persistía tras antibioticoterapia empírica durante su manejo ambulatorio. Como screening de FOD se procedió a la realización de pruebas laboratoriales y al barrido tomográfico donde se halló el pseudotumor pulmonar, que fue la principal pieza diagnóstica. En los resultados laboratoriales no se encontraron datos llamativos; incluyeron: valores normales o negativos, según corresponda, para hemograma (excepto por eosinofilia persistente de 25% en la fórmula relativa), hepatograma, urea, creatinina, PCR, VSG, ANA, Anti DNA, C3, C4, ANCA, rK39, HIV por Elisa, VHB y VHC, marcadores tumorales (CEA, Ca 125, Ca 19-9), policultivos y exámenes para tuberculosis (TB). Aspectos imagenológicos: Por tomografía axial computarizada (TAC) simple de tórax se informó tumoración pseudonodular de 35 mm en base pulmonar izquierda (Ver figura 1). En PET TC: hipercaptación en la zona del tumor referido. No se captaron adenopatías.
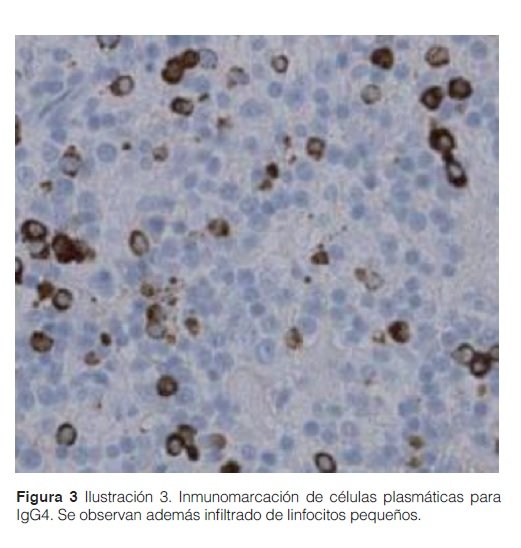
Se procedió a la resección del tumor por toracotomía. Se realizó tinción de inmuno-histoquímica en el espécimen resecado, cuyo informe fue “infiltrado mixto compuesto por linfocitos B y T con marcado aumento de células positivas para IgG4, sugerentes de neumonitis tipo IgG4 y fibrosis. Ausencia de granulomas” (Ver figura 2).
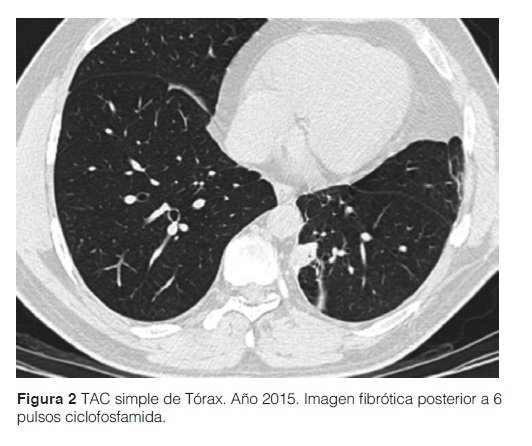
Ante este diagnóstico de ER-IgG4 el paciente recibe tratamiento con prednisona a dosis de 15 mg/día y azatioprina dosis ascendentes hasta 150 mg/día, sin respuesta favorable a los 12 meses. Se practica TAC contrastada de tórax 12 meses después de inicio de tratamiento, donde se encuentra una imagen nodular de similares características. Se procedió a rebiopsia de dicha imagen nodular donde se informa infiltrado intersticial linfoplasmocítico con invasión marcada de pared de vasos sanguíneos de mediano calibre; una segunda inmunohistoquímica informó resultados similares al anterior. Se inicia ciclofosfamida (CFM) ante diagnóstico de una probable vasculitis linfoplasmocítica y enfermedad pseudonodular por IgG4, con mejoría clínica. En la TAC torácica con contraste de control a los 6 meses del inicio de CFM se informa ausencia de nódulos pulmonares; en su lugar se observan finos trazos fibróticos con retracción de la pleura. (ver Figura 3)
DISCUSIÓN
El paciente comentado había presentado un cuadro de 5 años de evolución de pérdida de peso, FOD, que persistía tras antibioticoterapia empírica durante su manejo ambulatorio. Como screening de FOD se procedió al barrido tomográfico donde se halló el pseudotumor pulmonar, que fue la principal pieza diagnóstica.
La ER-IgG4 se caracteriza por la formación de pseudotumores con infiltrado linfoplasmocítico positivo para IgG4 en inmunohistoquímica, además de fibrosis estoriforme y a menudo elevación de IgG4 en suero1. Las manifestaciones clínicas dependen del órgano afectado, y en la mayoría de los casos coexisten síntomas constitucionales6. En nuestro caso no fue posible el dosaje sérico de IgG4 por problemas logísticos.
El diagnóstico definitivo de ER-IgG4 es por exclusión de otras patologías de características similares, sobre todo de carácter linfoproliferativo y cáncer de páncreas, seguido de infecciones como la tuberculosis, además de sarcoidosis, ciertos tipos de vasculitis, Síndrome de Sjögren, entre muchos otros6. Estas entidades fueron descartadas mediante los resultados de laboratorios de análisis clínicos e histológicos. Al mismo tiempo estos hallazgos acercaron la posibilidad diagnóstica de ER-IgG4, fundamentalmente por la inmunomarcación de IgG4 y la fibrosis en el material evaluado.
Los criterios diagnósticos han sido discutidos en el simposio internacional de ER-IgG4, celebrado en Boston, en el 2011 (Tabla 1). Fueron establecidos 3 pilares a saber: dominio clínico radiológico, serológico y sobre todo el histológico. Los hallazgos histológicos característicos incluyen infiltrado linfoplasmocítico con inmunomarcación para IgG4+, fibrosis con patrón estoriforme y flebitis obliterante7.

La relevancia de la serología es discutida por el hecho de que entre 3 y 30% de los pacientes con ER-IgG4 presenta valores normales de IgG4 en suero6. Sin embargo, existen datos sugerentes de que su dosaje podría relacionarse con la actividad de la enfermedad. La proporción de IgG4/IgG en el tejido biopsiado es de menor importancia para establecer el diagnóstico7.
Como dato adicional suele acompañarse con eosinofilia leve a moderada en sangre periférica, que fue el único hallazgo en el hemograma del paciente presentado8.
La ausencia de respuesta favorable al tratamiento inicial con prednisona y azatioprina, podría atribuirse a la dosis insuficiente, pues la dosis recomendada de prednisona es de 30 mg/día como pauta de inducción a la remisión, por lo menos durante un mes, para luego instalar un descenso gradual. No todos los pacientes requieren combinar corticoides con inmunosupresores, pero en nuestro caso fue necesario iniciar CFM, debido a la falta de buena respuesta. La terapia de mantenimiento se puede iniciar en pacientes con pobre respuesta o antecedente de recaídas; en este caso el paciente se encuentra actualmente con azatioprina a 150 mg/día. Otras opciones son micofenolato y rituximab6.
CONCLUSIÓN
De poco más de una década, la enfermedad relacionada a IgG4 (ER-IgG4) es una entidad clínico histológica emergente, que engloba varias enfermedades con descripciones poco frecuentes, que exige mantener una alta sospecha diagnóstica9.
Se estima que es una patología infra diagnosticada, en parte por desconocimiento y su reciente descripción y caracterización en la sociedad médica9.
En el 2008 se han publicado 63 casos, y para el 2013 el número de registros aumentó a 729, lo cual se relaciona con la unificación de criterios diagnósticos6. En la bibliografía consultada, no se encontraron publicaciones de casos de IgG4 en Paraguay, constituyéndose, éste, como el primer caso reportado. Se concluyó como diagnóstico de ER-IgG4 por la compatibilidad histológica y clínico radiológica, aun en ausencia de IgG4 sérica. El paciente presentó franca mejoría con el tratamiento instalado, actualmente sin reporte de recaída.
Se requiere más casos de ER-IgG4 para establecer características y pautas terapéuticas adecuadas a la población paraguaya.
BIBLIOGRAFÍA
(1) Stone JH, Zen Y, Deshpande V. IgG4-Related Disease. N Engl J Med. 2012;366(6):539–51.
(2) Erlij D, Ramos D, Montaña J, Kusnir P, Correa G, Neira O. Enfermedad relacionada a IgG4, el nuevo “gran simulador”: caso clínico. R ev Médica Chile. 2014;142(5):646–50.
(3) Carrillo Esper, R, Echeverría Vargas, JA. Enfermedad Relacionada con IgG4. Med Int Mex. 2013;29(1):53–61.
(4) Abud-Mendoza C. Enfermedades relacionadas con IgG4 (IgG4- R D), con horizonte no limitado a la enfermedad de Mikulicz. Reumatol Clínica. 2013;9(3):133–5.
(5) Chen LY, Wong PC, Noda S, Collins DR, Sreenivasan GM, Coupland RC. Polyclonal hyperviscosity syndrome in IgG4-related disease and associated conditions. Clin Case Rep. 2015;3(4): 217–26.
(6) Arezou Khosroshahi ZSW. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Amp Rheumatol Hoboken NJ. 2015;67(7):1688-99.
(7) Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol Off J U S Can Acad Pathol Inc. 2012;25(9):1181–92.
(8) Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH. IgG4- related disease. Annu Rev Pathol. 2014;9:315–47.
(9) Nares Martín E, Alvarez López F, Vargas Solís E, Araujo Ramirez O, Coss y Leon R, Sanchez Vargas AJ, et al. Enfermedad Relacionada a IgG4: una entidad multifacética. Rev Médica MD [Internet]. 2016 3):155-169.
Fecha de envío: 17/04/2016 - Fecha de aprobación: 20/05/2016