Revista Paraguaya de Reumatología, Vol. 2, N° 1, 2016
ISSN 2413-4341
ARTÍCULO ESPECIAL
PACIENTE CON ARTROPATÍA PROGRESIVA. UNA HISTORIA DIFERENTE. FORMAS ATENUADAS DE LA MUCOPOLISACARIDOSIS TIPO I. CASO CLÍNICO Y REVISIÓN DE LA LITERATURA
Imelda Martínez1, Pedro Babak2, Romina Sirtori3, Angeles Martínez4, Claudia Insfran4, Federico Rivarola5, Amalio Benítez6, Rosa Ayala7, Daniel Sánchez di Martino7
Miembros del equipo de trabajo para Mucopolisacaridosis Tipo I
1 Hospital de Clínicas y Hospital Central del Instituto de Previsión Social, Asunción, Paraguay
2 Servicio de Reumatología del Hospital Central del Instituto de Previsión Social, Asunción, Paraguay, Hospital Regional de Luque, Paraguay
3 Servicio de Imágenes del Hospital Central del Instituto de Previsión Social, Asunción, Paraguay
4 Servicio de Clínica Médica del Hospital Central del Hospital Central del Instituto de Previsión Social, Asunción, Paraguay y Urgencias del Hospital Nacional de Itaguá, Paraguay
5 Departamento de Otorrinolaringología del Hospital Central del Instituto de Previsión Social, Asunción, Paraguay
6 Departamento de Cardiología Pediátrica del Hospital Central del Instituto de Previsión Social, Asunción, Paraguay, Departamento de Cardiología Pediátrica, Hospital de Clínicas, Facultad de Ciencias Médicas de la Universidad Nacional de Asunción, Paraguay
7 Cátedra de Oftalmología, Hospital de Clínicas, Facultad de Ciencias Médicas de la Universidad Nacional de Asunción y Instituto de la Visión, Asunción, Paraguay
Autor para correspondencia. Correo electrónico: imeldanum@gmail.com (I. Martínez)
RESUMEN
Las mucopolisacaridosis son enfermedades de depósito lisosomal que se producen por una mutación en el gen que codifica la enzima lisosomal α-L-iduronidasa, necesaria para el catabolismo de los glicosaminoglicanos heparan sulfato y dermatan sulfato. La herencia es autosómica recesiva, con una prevalencia de 1-1,9/100.000 nacidos vivos. Es de carácter progresivo y multisistémico. Se han descripto tres fenotipos de la enfermedad, síndrome de Hurler, síndrome de Hurler-Scheie y síndrome de Scheie, según el grado de severidad de los síntomas y el compromiso neurológico que presenten. No existen parámetros clínicos claros que diferencien un fenotipo de otro, por lo que actualmente se acepta la diferenciación en fenotipo severo y fenotipo atenuado, teniendo en cuenta las opciones terapéuticas para cada forma de presentación. Los fenotipos atenuados tienen una presentación heterogénea en cuanto a la aparición de los rasgos toscos característicos, por lo que muchas veces pasan desapercibidas y esto contribuye al subdiagnóstico de las mismas. La artropatía está presente en la mayoría de los pacientes, es bilateral, simétrica y universal, afectando en mayor o menor medida a todas las articulaciones, causando restricción en la movilidad articular. Estos pacientes también presentan compromiso de vías aéreas altas, afectación cardiaca, oftalmológica y riesgo de mielopatía, que pueden ser causa de mayor morbilidad y mortalidad. El diagnóstico precoz es muy importante ya que la terapia de reemplazo enzimático con laronidasa mejora las manifestaciones clínicas no neurológicas y estabiliza la progresión de la enfermedad. Presentamos el caso clínico de una paciente con historia clínica de rigidez articular progresiva acompañado de disminución de la agudeza visual por opacidad de la córnea, infecciones otorrinolaringológicas de repetición, retraso en desarrollo (talla baja) y cirugía por hernia umbilical. Fue diagnosticada de Mucopolisacaridosis tipo I, fenotipo Scheie a los 21 años de edad, inició tratamiento con Laronidasa a los 23 años.
Palabras claves: Mucopolisacaridosis, Tipo I, fenotipos atenuados, laronisasa
PATIENT WITH A PROGRESSIVE ARTHROPATHY: A UNIQUE STORY. ATTENUATED FORMS OF MUCOPOLYSACHARIDOSIS TYPE I. CASE REPORT AND REVIEW OF THE LITERATURE.
ABSTRACT
Mucopolysaccharidosis type I are lisosomal storage diseases that are caused by a mutation on the gene that encodes the lisosomal enzyme α-L-iduronidase necessary for the glycosaminoglycan heparan sulfate and dermatan sulfate catabolism. The inheritance is autosomal recessive, with a prevalence of 1 to 1,9 per 100.000 live births. It is of progre-ssive and multisystemic nature. Three phenotypes of the diseases has been described, Hurler syndrome, Hurler-Scheie syndrome and Scheie syndrome, depending of the severity of the symptoms and the neurological impairment. There are not clinical parameters that differentiate one phenotype from the others. Therefore, currently it is accepted the difference between accentuated phenotypes and attenuated phenotypes taking into account the therapeutical options for each type. The coarse features onset in the attenuated phenotypes presents a heterogeneous display, this is the reason why it often goes unnoticed, contributing to its underdiagnoses. The arthropathy presented in most patients, is bilateral, symmetrical and universal, affecting to a greater or lesser extent to all joints, causing joint mobility restriction. These patients also present upper airway compromise, cardiac and ophthalmological involvement and risk of myelopathy, all these could lead to increased morbidity and mortality. Early diagnosis is very important, since enzyme replacement therapy with laronidase improves the non-neurological clinical manifestation and stabilizes the disease progression. We present a clinical case of a patient with a story of progressive joint mobility restriction, accompanied of decrease visual acuity due to corneal clouding, otorhinolaryngologic recurrent infections, developmental delay (short stature) and umbilical hernia surgery. Patient was diagnosed MPS I at 21 and started treatment with laronidase at 23 years old.
Keywords: Mucopolysaccharidosis type I, attenuated pheno-types, laronidase
INTRODUCCIÓN
Las mucopolisacaridosis son un grupo de patologías de depósito lisosoma1 que se caracterizan por la deficiente degradación de glicosaminoglicanos en distintos órganos y tejidos del cuerpo humano. Las manifestaciones mayores de la enfermedad dependen del tipo de enzima deficiente, es por ello que la presentación clínica y el tratamiento específico difieren para los varios tipos de mucopolisacaridosis. La mucopolisacaridosis tipo I (MPS I) es una enfermedad autosómica recesiva producida por una mutación en el gen que codifica la enzima lisosomal α-L-iduronidasa necesaria para la degradación de los glicosaminoglicanos (GAGs) heparán sulfato y dermatán sulfato. Estos GAGs se encuentran ampliamente distribuidos en el organismo, y es el motivo por el cual la MPS I es una enfermedad multisistémica y progresiva en cuanto a su evolución1. La incidencia global de todos los tipos de MPS es de 1/22.500, representando el 35% de las enfermedades de depósito lisosomal2, la incidencia de la MPS tipo I es de 0,99-1,99/100 000 nacidos vivos3.
Tradicionalmente fue clasificada en tres fenotipos distintos, dependiendo de la edad de inicio, velocidad de progresión y severidad de las manifestaciones neurológicas (síndrome de Hurler, síndrome de Hurler-Sheie, síndrome de Sheie, de mayor severidad a menor severidad respectivamente). Sin embargo hasta hoy día no existen criterios claros para la diferenciación de los distintos fenotipos4. Actualmente es aceptada la clasificación de formas tempranas severas de MPS I y formas atenuadas, esta distinción hace referencia a las distintas opciones terapéuticas disponibles para estos fenotipos diferenciados5. El acceso al tratamiento específico de los distintos fenotipos de MPS I hace necesario el diagnóstico precoz de manera a mejorar la calidad de vida de estos pacientes.
Describimos el caso de una paciente de 27 años de edad diagnosticada de MPS I fenotipo Scheie a los 21 años, el síntoma inicial fue la rigidez articular progresiva, que se inició a los 5 años, principalmente en manos, al que se añadieron manifestaciones como opacidad de la córnea e infecciones respiratorias de repetición, retardo del desarrollo con inteligencia normal.
CASO CLÍNICO
Paciente de sexo femenino de 27 años de edad (nacida en el año 1989), producto de una cuarta gestación planeada y deseada, parto, sin complicaciones. Peso al nacer: 4000 g. Es la cuarta hija de una pareja no consanguínea y sin historia de enfermedades genéticas en la familia.
Al nacer presentó un pequeño meningocele, que fue corregido quirúrgicamente y pies valgos. A los 3 meses se realizó un estudio de resonancia magnética nuclear del encéfalo que demostró secuela de meningocele, cisto de aracnoides, ausencia de hidrocefalia y cefalocele atrésico. Presentó un desarrollo psicomotor normal en los primeros años de vida, empezando a caminar a los 16 meses y a hablar alrededor de los 12 meses. La paciente completó la escuela primaria, la secundaria y la universitaria sin dificultades desde el punto de vista cognitivo. Entre los 5 y 6 años percibió leve rigidez en sus articulaciones. Los síntomas articulares fueron progresando, con mayor limitación funcional, por lo que la paciente realizaba fisioterapia frecuentemente con relativa mejoría. A los 8 años fue intervenida quirúrgicamente de una hernia umbilical. Desde la infancia presentaba infecciones otorrinolaringológicas a repetición.
Fue valorada por varios oftalmólogos por disminución de la agudeza visual y cambio de color de los ojos. En el año 2010 a los 21 años, tras una valoración por oftalmología fue remitida a un genetista, por sospecha de MPS. Fue derivada a un servicio especializado del Hospital de Clínicas del Brasil. Al examen clínico, presentaba buen estado general, macrocefalia, con facies infiltrada e hipertrofia gingival, peso de 46 Kg y talla de 131 cm (talla baja). La paciente presentaba opacidad corneana bilateral y se observaba al examen oftalmológico depósitos blanquecino/grisáceos polimorfos, localizados en forma difusa en todo el espesor del estroma corneal sin alteración del epitelio o endotelio, los cuales presentaban características normales. Además se evidenciaba un marcado aumento del espesor corneal en ambos ojos. A nivel de la retina no se evidenciaba alteración, las maculas estaban normales y el nervio óptico impresionaba normal con una excavación de 0,3 aproximadamente.
La auscultación cardiovascular fue normal, sin soplos. La ecocardiografía con doppler, informaba buena función sistólica y del segmento ventricular. Función diastólica preservada y una esclerosis de la válvula mitral y de la aortica. La insuficiencia valvular mitral era leve, sin dilatación ventricular izquierda. La insuficiencia valvular aórtica era leve a moderada. La presión pulmonar era normal. No se constató hepatoesplenomegalia. Las manos y los pies eran pequeños con una importante limitación de la movilidad articular y deformidad en flexión de los dedos. En la evaluación de la vía aérea superior se detectó un trastorno obstructivo de grado leve. Los estudios de laboratorio citoquímico, hemograma, perfil lipídico, glicemia, perfil renal, perfil hepático y tiroideo, fueron normales. Ante la sospecha de MPS se solicita la determinación de Glicosaminoglicanos en la orina, siendo el valor de 114 ul/ml. Se constató además una actividad de α-L-Iduronidasa disminuida en leucocitos de sangre periférica. Con estos resultados la paciente fue diagnósticada de MPS I.
Estudios radiográficos
Cráneo: Dolicocefalia. Engrosamiento de la bóveda craneal Silla turca en forma de ‟J”.
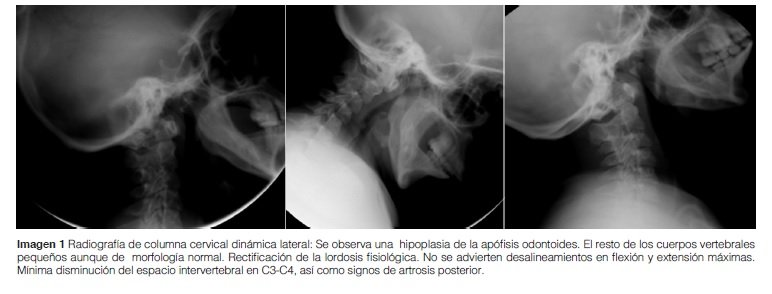
Columna cervical: Hipoplasia de la apófisis odontoides. El resto de los cuerpos vertebrales pequeños aunque de morfología normal (Imagen 1).
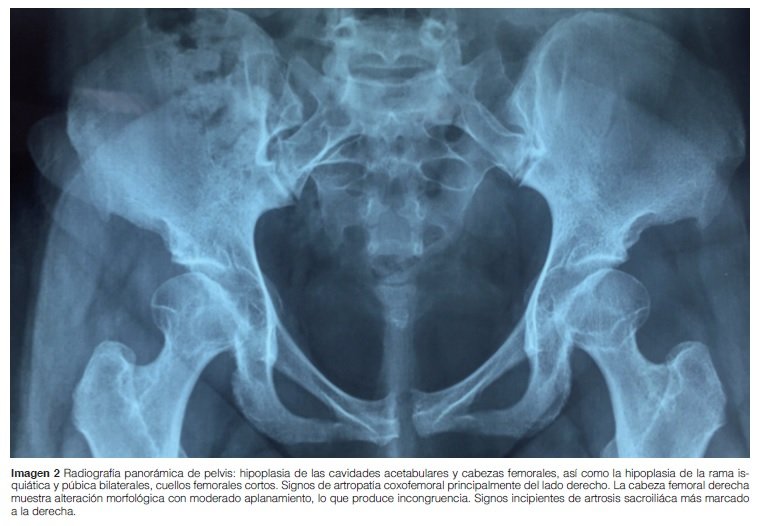
Caderas: Hipoplasia de las cavidades acetabulares y cabezas femorales, así como hipoplasia de la rama isquiática y púbica bilateralmente, los cuellos femorales cortos. La cabeza femoral derecha presenta alteración morfológica con moderado aplanamiento, lo que produce incongruencia (Imagen 2).
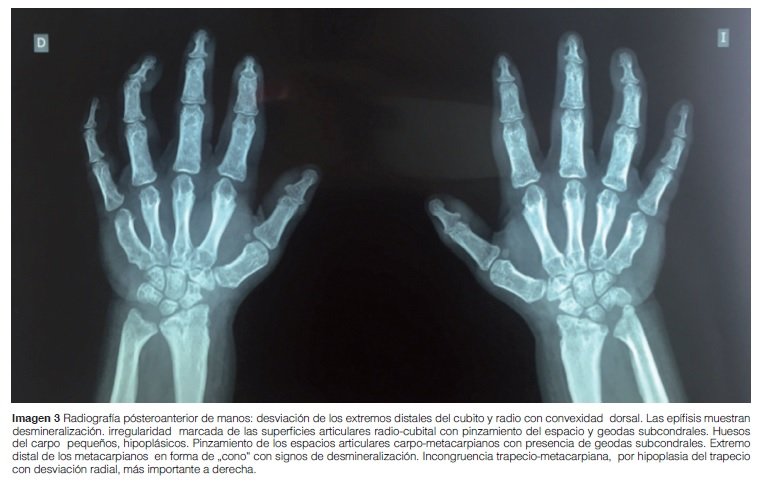
Manos: Desmineralización difusa, extremo distal de metacarpos en forma de cono, huesos del carpo hipoplásicos, pinzamiento y geodas subcondrales en espacios articulares carpo-metacarpianos (Imagen 3).
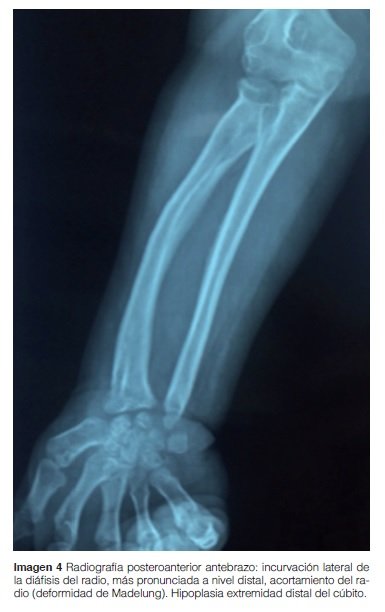
Antebrazo: Cúbito hipoplásico con curvatura del radio hacia cúbito más marcado a nivel distal (deformidad de Madelung) (Imagen 4).
En el año 2012, inició terapia de reemplazo enzimático con Laronidasa (Aldurazyme®), mostrando una franca mejoría, menos rigidez articular, por lo tanto mayor agilidad, piel más fina y tersa, ya no emite ronquidos al dormir, menos infecciones otorrinolaringológicas. En el 2014 por disminución de la agudeza visual por la opacidad de la córnea se indicó trasplante de córnea. En febrero de 2016 se realizó trasplante lamelar anterior profundo (DALK) en el ojo izquierdo sin complicaciones.
REVISIÓN DE LA LITERATURA
Patogenia
La MPS tipo I es el resultado de una mutación en el gen que codifica la enzima α-L-iduronidasa (IDUA), localizada en el cromosoma 4, de herencia autosómica recesiva. La IDUA se encarga de la degradación de los GAGs dermatán sulfato (DS) y heparán sulfato (HS), necesarias para mantener la homeostasis celular y el correcto crecimiento de los tejidos, dando como consecuencia la acumulación de DS y HS parcialmente degradados dentro de los lisosomas de varios tejidos, causando disfunción celular, tisular y orgánica progresivas, se acompaña de elevación de fragmentos de GAGs en sangre y orina1. El mecanismo fisiopatológico de daño no está bien dilucidado aún, además de la enfermedad de depósito en sí, existe una sobre regulación de varios genes y proteínas proinflamatorias, mayoritariamente citoquinas y proteasas. El resultado inicial de esta sobre regulación es la alteración estructural del tejido conectivo y cartilaginoso con empeoramiento de la elastogénesis, posteriormente aparece la enfermedad articular y ósea. La alteración en la degradación de los GAGs sería un gatillo inicial de una cascada de eventos que condicionaran el daño tisular multisistémico progresivo6.
Presentación clínica
Para los pacientes con fenotipos atenuados (síndrome de Hurler-Scheie, síndrome Scheie) los primeros síntomas aparecen entre los 3-7 años y 5-13 años respectivamente7.
Según datos del registro (MPS I Registry) los síntomas más frecuentemente reportados en los pacientes con síndrome Hurler-Scheie fueron: deterioro cognitivo, que se inició entre los 3-4 años, acompañado de desarrollo de rasgos faciales toscos y hernias (umbilical e inguinal), posteriormente se presentaron macroglosia, trastornos del sueño y ronquidos, contracturas articulares y disostosis múltiple. Las opacidades corneales y la hepatoesplenomegalia aparecieron entre los 4-5 años. Las manifestaciones que aparecieron más tardíamente en estos pacientes fueron las alte-raciones en válvulas cardiacas, displasia de cadera y síndrome del túnel carpiano, con una media de edad de inicio de 5,7, 6,2 y 7,4 años respectivamente7.
Para los pacientes con síndrome de Scheie, uno de los primeros síntomas registrados fueron las hernias, con una media de edad de inicio de 7,6 años, las contracturas articulares fueron reportadas con una media de edad de 8 años, la aparición de displasia de cadera fue reportada con una media de edad de 8,4 años. Posteriormente aparecieron las alteraciones del sueño y ronquidos junto con el desarrollo de rasgos faciales toscos y hepatomegalia. Los síntomas reportados como más tardíos fueron las opacidades corneales, esplenomegalia, alteraciones de válvulas cardiacas y el síndrome del túnel carpiano7.
Apariencia física y craneofacial
Los pacientes con formas atenuadas de MPS I pueden presentar gran variabilidad fenotípica. Los rasgos faciales toscos son menos llamativos que en las formas severas. Estos rasgos incluyen cuello corto, mandíbula cuadrada y micrognatia5.
Manifestaciones esqueléticas
Son las que contribuyen mayoritariamente a la mala calidad de vida de estos pacientes.
Disostosis múltiple: es la constelación de las anormalidades esqueléticas clásicamente vistas en las MPS, son el resultado del defecto en el crecimiento óseo endocondral y membranoso. La cifosis con o sin escoliosis es el resultado de la hipoplasia de los cuerpos vertebrales. La superficie acetabular es poco profunda y amplia, permitiendo subluxación o luxación franca de la cabeza femoral. Las rodillas están típicamente en valgo8.
A nivel axial también se observa ampliación de las dimensiones del cráneo, aumento del espesor de la bóveda craneal, silla turca en forma de J, hipoplasia de la apófisis odontoides, que puede condicionar inestabilidad cervical. Las clavículas y las costillas son más grandes. En los huesos largos las epífisis son hipoplásicas y las diáfisis están engrosadas, dando una apariencia tosca. El radio distal a menudo se curva hacia un cúbito hipoplásico (deformidad de Madelung).
Los metacarpianos son cortos, con adelgazamiento proximal dando un aspecto de bala8.
Las contracturas articulares están descriptas en el 87,7% de los pacientes con síndrome de Scheie, frecuentemente asociada a otras manifestaciones osteoarticulares9. Son bilaterales y simétricas, sin signos inflamatorios locales, principalmente en articulaciones de las manos y hombros. Se presenta como una artropatía progresiva, en su evolución condiciona restricción universal de la movilidad de todas las articulaciones5. En las formas atenuadas generalmente es el primer síntoma que deriva en atención médica8,9. En muchos pacientes con síndrome de Scheie las típicas características toscas pueden estar ausentes y es por ello que muchas veces antes del diagnóstico de MPS tipo I son diagnosticados de otras patologías más prevalentes, entre ellas: esclerodermia, distrofia muscular, artritis idiopática juvenil, artritis reumatoide, enfermedad de Perthes, osteogénesis imperfecta, dolores del crecimiento, dermatomiositis, polimiositis y fiebre reumática10,11,12.
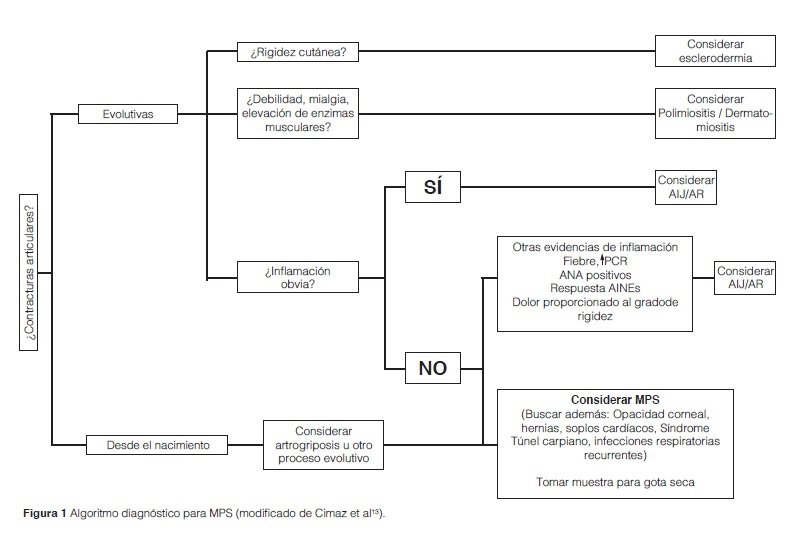
Teniendo en cuenta la baja prevalencia de la enfermedad y el subdiagnóstico de la misma se han diseñado algoritmos diagnósticos enfocados a incluir a la MPS dentro del diagnóstico diferencial de las artropatías (Figura 1)13,14.
Síndrome del túnel del carpo: reportado en 66,7% de los pacientes con fenotipos atenuados, con una media de edad de inicio de 13,1 años9. El 58,3% de los síndromes del túnel del carpo iniciados en la infancia están relacionados a MPS, el síndrome del túnel del carpo no es una patología frecuente de la infancia y generalmente se debe a una enfermedad subyacente15.
Talla baja: varios estudios fueron realizados en modelos animales, estos estudios revelaron que existe una alteración primaria morfogénica en el desarrollo tanto de la placa de crecimiento como de la estructura del hueso cortical16. El componente endocrinológico también está asociado al fallo en la adquisición de la talla esperada, se describieron alteraciones en la función de las glándulas pituitarias, tiroides, testículos y ovarios8. Los pacientes con fenotipo atenuado presentan un grado variable de retardo del crecimiento, pudiendo incluso alcanzar la talla esperada para la edad, pero no lograr la talla genética esperada.
Manifestaciones oftalmológicas
Las opacidades corneales son manifestaciones frecuentes en los pacientes con fenotipos atenuados, con una media de edad de inicio de 9,1 años9. Las alteraciones oculares se observan en todos los tipos de MPS ocasionando frecuentemente una importante disminución de la visión. A nivel de la córnea existe una opacidad progresiva que afecta todo el espesor del estroma corneal y típicamente se describe como vidrio esmerilado y es debido el depósito de GAGs. Este depósito se localiza intra y extracelularmente en las estructuras corneales. Inicialmente se presenta como una opacidad punctata fina periférica del estroma anterior corneal que más tarde progresa y se vuelve difusa, resultando una pérdida de la agudeza visual17,18.
En la MPS I fenotipo Scheie se observan numerosas vacuolas que contienen material fibrilogranular en las células epiteliales, queratocitos y células endoteliales. La membrana basal epitelial contiene frecuentes rupturas y ondulaciones. La capa de Bowman está muy atenuada. Las fibras de colágeno son irregulares, se encuentran elongadas y espaciadas. La membrana de Descemet impresiona ser normal19. Estas alteraciones resultan en una en la reducción de la transparencia corneal y aumento de la dispersión de la luz20.
Además de la afectación de la córnea los pacientes con fenotipos atenuados también pueden presentar glaucoma, degeneración retiniana y atrofia del nervio óptico21,22.
Presencia de hernias
La matriz extracelular está compuesta por fibras elásticas, colágeno y GAGs. La función de los GAGs como componente de la matriz extracelular además de proveer soporte estructural a la red de fibras elásticas y colágeno también juega un rol importante en el ensamblaje de fibras elásticas y microfillas23. Antecedentes de hernias fueron reportados en un 61,5% de los pacientes con fenotipos atenuados, con una media de edad de 3,3 años, la más frecuente es la hernia umbilical seguida de hernia inguinal, derecha o izquierda9. Según datos del registro (MPS I Registry) el 40% y 36% de los pacientes con sindrome de Hurler-Scheie y sindrome de Scheie respectivamente reportaron antecedentes de cirugías de reparación de hernias, incluso antes del diagnóstico24.
Manifestaciones otorrinolaringológico
Los pacientes con MPS presentan problemas respiratorios de vía aérea alta y baja. La acumulación de GAGs causa: macroglosia, engrosamiento de la mucosa de la faringe, laringe y todo el árbol traqueobronquial, con disminución de la luz. También puede presentarse estenosis traqueal por depósitos de GAGs en el cartílago tra-quobronquial. Estas complicaciones hacen que exista un claro aumento de la morbilidad y mortalidad de estos pacientes25. Las manifestaciones de vía aérea alta se inician en etapas tempranas de la enfermedad en todos los fenotipos de MPS I, los pacientes son frecuentemente referidos al Otorrinolaringólogos antes del diagnóstico. En los fenotipos atenuados se han reportado macroglosia, aumento del tamaño de las amígdalas, hiperreactividad de la vía aérea, asma y alteraciones del sueño, estos pacientes también precisaron en un alto porcentaje cirugías relacionadas, incluso antes del diagnóstico de MPS9,26.
Manifestaciones cardiovasculares
La complicación cardiaca más frecuente en la MPS I es la afectación valvular, reportada en el 87,9% de los pacientes con una media de edad de inicio de 11,7 años9. En orden de frecuencia se pueden afectar las válvulas mitral, aórtica, pulmonar y tricuspídea, el compromiso es mayormente del tipo insuficiencia que estenosis, con alta frecuencia de reemplazo valvular. También se han reportado estrechamiento y oclusión coronaria, estenosis aorta torácica y aorta abdominal, HTA secundaria a estenosis aórtica, dilatación de la aorta ascendente. La afectación cardiovascular mucha veces permanece silente, no obstante es una causa importante de mortalidad temprana en estos pacientes27,28.
Manifestaciones neurológicas
En los fenotipos atenuados el deterioro cognitivo es leve o está ausente. La manifestación neurológica más frecuentemente descripta en estos pacientes es la mielopatía, el canal medular óseo suele ser normal, sin embargo puede existir un engrosamiento de la duramadre por los depósitos de GAGs, existen reportados casos donde la mielopa-tía es una manifestación inicial de la MPS29. También se pueden desarrollar quistes aracnoideos5.
Diagnóstico
El diagnóstico precoz mejora los resultados en el tratamiento de los pacientes con MPS I, sin embargo dado que las manifestaciones iniciales no son específicas y son comunes a otras patologías más frecuentes el diagnóstico de estos pacientes muchas veces se ve retra sado por varios años. Según resultados del registro de MPS (MPS I Registry) el retraso en el fenotipo Hurler-Scheie oscila entre 0,9 y 9,3 años, en el fenotipo Scheie oscila entre 1,5 y 47,3 años, en ambos casos, muchas veces ya cuando existe un daño orgánico irreversible30.
Para el diagnóstico confirmatorio de la MPS tipo I es necesario demostrar la deficiente actividad de la enzima IDUA, esta determinación puede ser realizada en leucocitos, fibroblastos, plasma. El test de gota de sangre seca es un método que ofrece muchas ventajas, no es invasiva y es más fácil de transportar. Una actividad enzimática < 10% del valor de referencia es compatible con el diagnóstico de MPS I, aunque cada laboratorio puede establecer sus propios valores de referencia, no obstante ante un resultado normal con alta sospecha hay que recurrir a los métodos clásicos de diagnóstico31,32.
El estudio confirmatorio del genotipo no es necesario de manera habitual. Está indicado en los estudios pre y post natales, para estudios de consejería genética y en estudios clínicos. Hay una correlación genotipo-fenotipo de hasta el 70%5.
El análisis de GAGs en orina mediante reacción colorimétrica o cromatografía puede ser un test que apoye el diagnóstico, no obstante siempre se va requerir del estudio de la actividad enzimática de la enzima IDUA31,32.
TRATAMIENTO
Para el cuidado de los pacientes con MPS I se requieren evaluaciones regulares, cuidados de soporte y tratamiento de las manifestaciones sistémicas. Debido a la complejidad y poca frecuencia de esta patología los pacientes deben ser evaluados por equipos multidisciplinarios mejorando así la experiencia en MPS de todo el equipo, para un mayor beneficio de los pacientes. Históricamente los pacientes con MPS se beneficiaban solo de tratamientos paliativos. En 1980 fue realizado el primer trasplante alogénico de células madre hematopoyéticas con éxito en un niño de un año de edad33.
La terapia de reemplazo enzimático (TRE) con larónidasa (enzima humana recombinante IDUA, Aldurazyme) es el tratamiento primario de elección para pacientes con formas atenuadas de la enfermedad, producida por tecnología DNA recombinante y aprobadas para utilización en el año 2003 por FDA y EMA34,35.
Laronidasa no cruza la barrera hematoencefálica, su administración es segura, mejora las manifestaciones clínicas no neurológicas y estabiliza la progresión de la enfermedad, sin embargo se obtienen mejores resultados a largo plazo cuando se inicia la terapia en etapas precoces36,37.
CONCLUSIÓN:
Actualmente el diagnóstico precoz de la mucopolisacaridosis es una prioridad en los pacientes que la padecen, ya que existe una terapia de reemplazo enzimático específica para la enfermedad que ha demostrado marcados beneficios, sobre todo cuando es iniciada en etapas tempranas, antes de la instauración del daño de órgano irreversible.
Es muy importante que el reumatólogo tenga en cuenta a esta patología como causante de artropatía progresiva y la incluya dentro de sus diagnósticos diferenciales, apoyado en el cuadro clínico del paciente, principalmente cuando va asociado a opacidad de la córnea y antecedente de hernia umbilical o inguinal para los fenotipos atenuados.
Dada la naturaleza multisistémica de la enfermedad estos pacientes deben ser valorados por un equipo multidisciplinario conformado por internistas, pediatras, cardiólogos, reumatólogos, ortopedistas, neurólogos, otorrinolaringólogos, radiólogos y oftalmólogos
BIBLIOGRAFÍA
(1) Derbis Campos D, Monaga M. Mucopolysaccharidosis type I: current knowledge on its pathophysiological mechanisms. Metab Brain Dis. 2012; 27:121-129.
(2) Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249-54.
(3) Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, Niezen-Koning KE, van Diggelen OP. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105:151-6.
(4) De Ru M, Teunissen Q G, van der Lee J et al. Capturing phenotypic heterogeneity in MPS I: results of an international consensus procedure. Orphanet Journal of Rare Diseases. 2012;7:22.
(5) GeneReviews® [Internet]. Mucopolysaccharidosis Type I (October 31, 2002; Last Update: February 11, 2016). Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK1162/
(6) Villani G RD, Chierchia A, Di Napoli D, Di Natale P. Unfolded protein response is not activated in the mucopolysaccharidoses but protein disulfide isomerase 5 is deregulated. J Inherit Metab Dis. 2012;35(3):479-93.
(7) Beck M, Arn P, Giugliani R. The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med. 2014;16(10):759-
(8) White K K. Orthopaedic aspects of mucopolysaccharidoses. Rheumatology.2011;50:26-33.
(9) Thomas JA, Beck M, Clarke JT, Cox GF. Childhood onset of Scheie syndrome, the attenuated form of mucopolysaccharidosis I. J Inherit Metab Dis. 2010;33:421-7.
(10) Melikoglu MA, Kocabas H, Sezer I, et al. Legg–Perthes diseaselike joint involvement and diagnosis delay in Scheie syndrome: a case report. Clin Rheumatol. 2007;26:1937-1939.
(11) Cimaz R, Vijay S, Haase C, et al. Attenuated type mucopolysaccharidosis in the differencial diagnosis of juvenile idiopathic arthritis: a series of 13 patients with Scheie syndrome. Pediatric Rheumatology. 2006;24:196-202.
(12) Raiman J, D’Aco K. An 8-Year-Old Girl with a History of Stiff and Painful Joints. Pediatric Annals. 2014;43(3):307-309.
(13) Cimaz R, Coppa GB, Koné-Paut I et al. Joint contractures in the absence of inflammation may indicate mucopolysaccharidosis. Pediatric Rheumatology. 2009;18(7):1-8.
(14) Lehman TJ, Miller N, Norquist B, et al. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50:41-48.
(15) Van der Meir N, De Smet L. Carpal tunnel syndrome in children. Journal of Pediatric Orthopedics. 2005;14:42-45.
(16) C lark LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology. 2011;50:13-18.
(17) Ashworth J, Biswas S, Wraith E, Lloyd C.: Mucopolysaccharidoses and the Eye Survophthal. 2006;51:1-17.
(18) Rosen DA, Haust MD, Yamashita T et al: Keratoplasty and electron microscopy of the cornea in systemic mucopolysaccharidosis (Hurler‘s disease). Can J Ophthalmol. 1968;3:218.
(19) Zabel R, Mac Donald I, Mintsioul is G, Addison D: Scheie´s Syndrome. An ultrastuctural analysis of the cornea. Ophthalmology. 1989;96:1631-38.
(20) Laver NM, Friedlander MH, McLean IW: Mild form of Maroteaux Lamy síndrome: corneal histopathology and ultrastructure. Cornea. 1998; 17:664-8.
(21) Summers CG, Ashworth JL. Ocular manifestations as key features for diagnosing mucopolysaccharidoses. Rheumatology. 2011;50: 34 40.
(22) Quigley HA, Maumenee AE, Stark WJ. Acute glaucoma in systemic mucopolysaccharidosis I-S. Am J Ophthalmol. 1975; 80(1):70-2.
(23) Barnett C, Langer JC, Hinek A et al. Looking past the lump: genetic aspects of inguinal hernia in children. Journal of Pediatric Surgery. 2009;44,1423-143.
(24) Arn P, Wraith JE, Underhill L. Characterization of Surgical Procedures in Patients with Mucopolysaccharidosis Type I: Findings from the MPS I Registry. J Pediatr. 2009;154:859-64.
(25) Morimoto N, Kitamura M, Kosuga M, Okuyama T. CT and endoscopic evaluation of larynx and trachea in mucopolysaccharidoses. Molecular Genetics and Metabolism. 2014;112:154-159.
(26) Arn P, Bruce L, Wraith JE. Airway-Related Symptoms and Surgeries in Patients with Mucopolysaccharidosis I. Annals of Otology, Rhinology & Laryngology. 2014;1-8.
(27) Braunlin EA, Harmatz PR, Scarpa M. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011:34:1183-1197.
(28) Soliman OI, Timmermans RG, Nemes A et al. Cardiac abnormalities in adults with the attenuated form of mucopolysaccharidosis type I. J Inherit Metab Dis. (2007)30:750-757.
(29) Furukawa Y, Hamaguchi A, Nozaki I, et al. Cervical pachymeningeal hypertrophy as the initial and cardinal manifestation of muco polysaccharidosis type I in monozygotic twins with a novel mutation in the alpha-l-iduronidase gene. Journal of the Neurological Sciences. 2011;302:121-125.
(30) Pastores G, Arn P, Beck M et al. The MPS I registry: design, methodology, and early findings of a global disease registry for monitoring patients with mucopolysaccharidosis type I. Mol Genet Metab. 2007;91:37-47.
(31) Bay L, Amartino A, Barreiro C y col. Consenso de diagnóstico y tratamiento de la mucopolisacaridosis de tipo I. Arch Argent Pediatr. 2008;106(4):361-368-361.
(32) Martins AM, Dualibi AP, Norato D et al. Guidelines for the Management of Mucopolysaccharidosis Type I. The Journal of Pediatrics. 2009;55(4)suppl 2.
(33) Muenzer J. Overview of the mucopolysaccharidoses. Rheumatology. 2011;50:4-12.
(34) United States Food and Drug Administration. Aldurazyme approval information. Available at, http://www.fda.gov/cder/biologics/products/ larobio043003.htm;2003.
(35) European Agency for the Evaluation of Medical Products. Aldurazyme approval information. Available at, http://www.emea.eu.int/humandocs/Humans/EPAR/aldurazyme/aldu azyme.htm;2003.
(36) Sannaa A, Bay L, Barbouth D et al. Early treatment with laronidase improves clinical outcomes in patients with attenuated MPS I: a retrospective case series analysis of nine sibships. Orphanet Journal of Rare Diseases. 2015;10:131.
(37) Gabrielli O, Clarke LA, Ficcadenti A. 12 year follow up of enzymereplacement therapy in two siblings with attenuated mucopolysaccharidosis I: the important role of early treatment. BMC Medical Genetics. 2016;17:19.
Fecha de envío: 01/04/2016 - Fecha de aprobación: 15/05/2016