Revista Paraguaya de Reumatología, Vol. 1, Nº 1, 2015
ISSN 2413-4341
CASO CLÍNICO
SÍNDROME ANTIFOSFOLÍPIDO EN UN VARÓN CON ARTRITIS REUMATOIDEA
Beatriz Di Martino1, Rosalba Riveros1, Tatiana Moreno1, Mirtha Rodríguez1, Oilda Knopfelmacher1, Lourdes Bolla1
1Cátedra de Dermatología, Hospital de Clínicas, Facultad de Ciencias Médicas, Universidad Nacional de Asunción, Paraguay
Autor para correspondencia. Correo electrónico: beatrizdimartino@gmail.com(B. Di Martino)
RESUMEN
El Síndrome antifosfolípido (SAF) es una patología autoinmune que se caracteriza por presentar trombosis vascular, abortos recurrentes, así como una elevación persistente de los anticuerpos antifosfolípidos. Desde su primera descripción en el año 1983, una amplia variedad de manifestaciones dermatológicas han sido asociadas a este síndrome, siendo las más frecuentes el livedo reticularis y las úlceras cutáneas. La presencia de los anticuerpos antifosfolípidos ha sido descrita en diversas patologías como en enfermedades autoinmunes, infecciones, neoplasias y de forma secundaria al uso de determinados fármacos. Presentamos el caso de un varón de 72 años de edad con diagnóstico de Artritis Reumatoide y SAF secundario, por su relacion inusual.
Palabras claves: Síndrome antifosfolípido, Artritis Reumatoidea, Anticuerpos antifosfolipidos
ANTIPHOSPHOLIPID SYNDROME IN A MALE PATIENT WITH RHEUMATOID ARTHRITIS.
ABSTRACT
The antiphospholipid syndrome (APS) is an autoimmune disease characterized by vascular thrombosis, recurrent abortions and persistent elevation of antiphospholipid antibodies. Since its first description in 1983, a wide variety of dermatological manifestations have been associated with this syndrome, most frequently livedo reticularis and skin ulcers. The presence of antiphospholipid antibodies has been described in several diseases such as autoimmune diseases, infections, neoplasms and secondarily to the use of certain drugs. We report the case of a 72-year-old diagnosed with Rheumatoid Arthritis and secondary APS, for its unusual relationship.
Keywords: Antiphospholipid syndrome, rheumatoid arthritis, antiphospholipid antibodies
INTRODUCCIÓN
El Síndrome Antifosfolípido (SAF) es una patología autoinmune que se caracteriza por la presencia de anticuerpos antifosfolípidos (AAF), fenómenos trombóticos (arteriales y/o venosos) y/o pérdidas fetales recurrentes.(1) En su inicio fue asociado con el Lupus Eritematoso Sistémico (LES) y posteriormente se asoció con otros trastornos autoinmunes pero en un porcentaje mucho menor2. A continuación se presenta el caso de un paciente con diagnóstico de Artritis Reumatoide (AR) y SAF secundario.
Caso Clínico: varón de 72 años de edad con antecedente de AR, de procedencia rural. Refiere enrojecimiento del 1/3 inferior de ambos miembros inferiores (MMII), de predominio izquierdo, de 1 mes de evolución. A las dos semanas se agrega ennegrecimiento y secreción purulenta de las lesiones por lo que decide consultar en un centro asistencial de su localidad. En dicho centro es evaluado y diagnosticado de gangrena húmeda, por lo que se realiza una amputación supracondílea izquierda. Posterior al procedimiento es remitido al Hospital de Clínicas de la Facultad de Ciencias Médicas de la Universidad Nacional de Asunción para optimizar el manejo clínico. A los 5 días de su internación en nuestro centro, se objetivó un empeoramiento de las lesiones rojizas en el miembro inferior derecho, las cuales se tornan negruzcas, sobre todo en dedos y planta del pie, por lo cual solicitan evaluación por nuestro departamento. El paciente presenta antecedentes de Hipertensión arterial, cardiopatía isquémica, secuelas de accidente cerebro vascular isquémico, AR desde hace varios años, Síndrome de Cushing iatrogénico, en tratamiento con enalapril 20mg cada 12 horas, automedicado con corticoides en forma irregular; actualmente con sepsis a punto de partida de piel y partes blandas, tras amputación del miembro inferior izquierdo 15 días antes del ingreso.
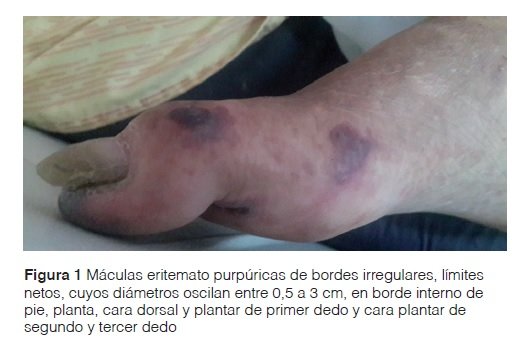
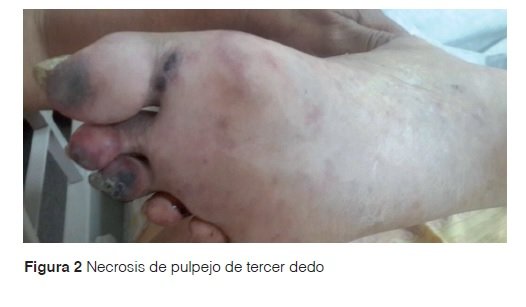
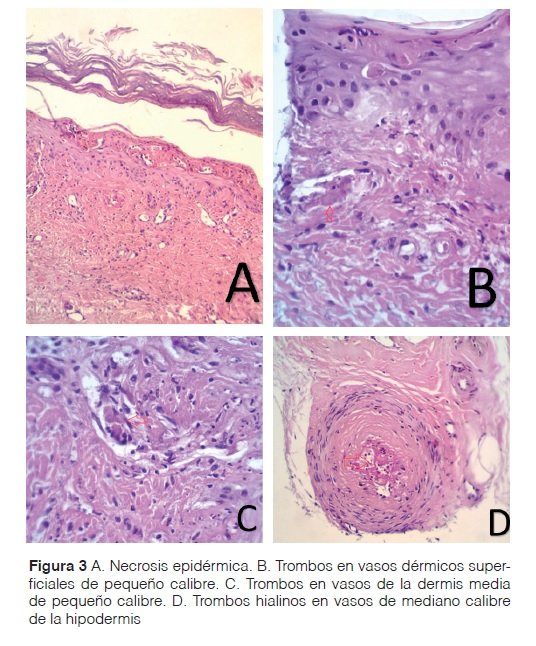
Examen físico: paciente caquéctico, con facies de luna llena, presencia de máculas eritemato purpúricas de bordes irregulares y límites netos, cuyos diámetros oscilaban entre 0,5 y 3 cm. Estas lesiones se ubicaban en el borde interno y planta del pie derecho, en la cara dorsal y plantar de primer dedo y en la cara plantar del segundo y tercer dedo (Figura 1). En el pulpejo del tercer dedo del pie derecho, se objetivó la presencia de necrosis (Figura 2). Al evaluar la presencia de pulso, se constató un pulso pedio disminuido. El estudio anatomopatológico de las lesiones describió la presencia de necrosis de las capas epidérmicas superiores (Figura 3A), presencia de material hialino trombótico en vasos de pequeño calibre de la dermis superficial (Figura 3B) y media (Figura 3C) y en vasos profundos de mediano calibre (Figura 3D).
Laboratorio: Leucocitosis de 18.000/mm3 con 86% de neutrófilos, Hb: 9,1gr/dl, Plaquetas: 220.000/mm3, TSH: <0,01uUI/ml (VN: 0,4-4), TP: 80%, PCR 50 (+), VSG 80mm, ANA negativo, FR + 196 (VN <12APL)C3: 6,5 (15-33), C4: 44,3 (82-150), Anticuerpos anticardiolipina: IgA (+) 266 APL/ml (VN <18APL/ml), IgM (+) 486 MPL/ml (VN <18MPL/ml), IgG (+) 245 GPL/ml (VN <18GPL/ml), Anticoagulante lúpico: (+) 110U/ml (VN <18U/ml)
Con dichos resultados se concluye diagnóstico de SAF en paciente con AR. Recibió tratamiento antibiótico por 10 días con Ceftazidima y Clindamicina para el control del cuadro séptico, más anticoagulación, con buena evolución clínica.
Discusión
El SAF es una entidad multisistémica caracterizada por la presencia persistente y elevada de anticuerpos antifosfolípidos, trombosis arterial y/o venosa, y/o a-bortos espontáneos recurrentes. Se clasifica en SAF primario sin enfermedad subyacente y SAF secundario a diversas patologías ya sean autoinmunitarias o no (enfermedades oncológicas, infecciosas, vasculopatías, hemopatías y medicamentos) donde la enfermedad que se asocia en forma más frecuente es el LES1-2.
Los anticuerpos antifosfolípidos (AAF) son un grupo de inmunoglobulinas IgG, IgM e IgA que están dirigidos, contra diferentes complejos formados por los fosfolípidos aniónicos de las membranas celulares (principalmente cardiolipina) y proteínas plasmáticas, que actúan como cofactores. Inicialmente, se consideraba que estos anticuerpos reaccionaban exclusivamente con los fosfolípidos; sin embargo, con el uso de antígenos más puros en las pruebas de laboratorio, se ha determinado que el blanco de estos autoanticuerpos puede ser un complejo fosfolípido-proteína o simplemente una proteína (anticuerpo anticofactor).
El cofactor de mayor relevancia actualmente es la b2-glicoproteína I (b2GP-I), pero también se citan a la protrombina, la proteína C, la proteína S y la anexina V, entre otros2-4.
Estos anticuerpos provocan la aparición de trombosis, alterando la función que estas proteínas tienen en el proceso de la coagulación, además de actuar directamente sobre el endotelio vascular y el sistema inmune.(2,5)
El SAF fue descrito por primera vez por Hughes en el año 1983, siendo las principales manifestaciones clínicas: la trombosis venosa profunda (28-55%), la trombosis arterial principalmente a nivel cerebral, y las anomalías cardíacas valvulares con embolismos periféricos. Otras manifestaciones del SAF incluyen: trombocitopenia (40-50%), anemia hemolítica (14-23%) y la presencia de livedo reticularis (11-23%). Aunque la manifestación renal es muy frecuente en el LES, es apenas reconocida como parte del SAF y en aquellos que lo presentan invariablemente se asocia con hipertensión arterial. El 20% de las mujeres con pérdidas fetales recurrentes presenta SAF, y éstas ocurren más típicamente durante el segundo trimestre del embarazo2,6-8.
Las lesiones cutáneas asociadas al SAF son frecuentes y pueden ser signos de presentación de la enfermedad en el 41% de los casos, siendo la necrosis cutánea extendida la más rara y severa. El livedo reticularis es la manifestación más común e indica para algunos autores, diagnóstico de presunción, apareciendo como consecuencia de la estasis sanguínea en los sistemas superficiales de drenaje de la piel y está probablemente relacionada a la oclusión vascular observada en el SAF, presente en el 55% de los pacientes con SAF primario. Puede observarse también en enfermedades infecciosas y autoinmunitarias, particularmente en el LES, aun en ausencia de anticuerpos antifosfolípidos. Sin embargo, la presencia de livedo reticularis moderado a severo en el paciente lúpico se asocia significativamente con anticuerpos anticardiolipina elevados. Livedo racemosa es otra forma de livedo caracterizado por un patrón violáceo llamativo en forma de red que se diferencia del livedo reticularis por ser generalizado, apareciendo no solo en los miembros inferiores sino también en el tronco y glúteos. Es un signo típico del Síndrome de Sneddon, pero se puede observar además en el LES con o sin SAF, en la trombocitemia esencial, policitemia vera y poliarteritis nodosa. Se asocia frecuentemente con eventos cerebrovasculares, trombosis arteriales, morbilidad durante el embarazo y es considerado un factor de riesgo trombótico independiente y adicional por lo que es importante diferenciar las 2 formas de livedo1,6,9,10.
En cuanto a los trastornos asociados al SAF se citan:
Enfermedades inmunológicas: LES (25-50%), Púrpura Trombocitopénica Autoinmune, Artritis Reumatoide, Artritis Psoriásica, Síndrome de Sj�gren, Arteritis de células gigantes, Polimialgia reumática, Enfermedad Mixta del Tejido Conjuntivo, Esclerosis Sistémica, Enfermedad de Behcet, Poliarteritis Nodosa, Dermatomiositis/polimiositis, Anemia Hemolítica Autoinmune y Hepatitis crónica activa, vasculitis autoinmunes, Esclerodermia sistémica.
Neoplasias: tumores sólidos, leucemias, desórdenes linfoproliferativos/enfermedad de Hodgkin, mieloma múltiple y micosis sistémica. Enfermedades hematológicas: enfermedad de von Willebrand, mielofibrosis y paraproteinemias.
Enfermedades infecciosas: sífilis, enfermedad de Hansen, tuberculosis, micoplasma, enfermedad de Lyme, malaria, sida, hepatitis A y C, mononucleosis, adenovirosis, parvovirosis, sarampión, varicela e infecciones bacterianas (endocarditis y sepsis).
Enfermedades neurológicas: miastenia gravis, esclerosis múltiple y jaqueca (hemicránea).
Medicamentos: clorpromazina, fenitoína, hidralazina, procainamida, quinidina, estreptomicina y fenotiazina(9-11)

El diagnóstico del SAF se realiza en base a criterios establecidos en el Octavo Congreso Internacional de Sapporo en 1999, y a su vez revisados tanto en el año 2004 en Sidney como en el año 2006 en el Consenso Internacional de SAF2,6,12-13 y se citan a continuación:
Se evitará el diagnóstico de SAF cuando existan menos de 12 semanas o más de 5 años de intervalo entre la positividad de los Anticuerpos Antifosfolípidos (AcAF) y las manifestaciones clínicas2,4
Nuestro paciente presentaba AcAF positivos en una ocasión y trombosis de pequeños vasos certificados por histología sin inflamación de la pared vascular. Queda pendiente recibir los resultados analíticos de los segundos anticuerpos, que aún no se han recibido debido a que el paciente aún no ha regresado a control.
Para hablar de tratamiento de SAF es necesario tener en cuenta las diversas formas clínicas que pueden presentarse y de la presencia de enfermedad sistémica subyacente. Los corticoides sistémicos u otros agentes inmunosupresores están indicados en la forma secundaria para tratar la enfermedad asociada. La profilaxis y el tratamiento de trombosis se efectúan con agentes anticoagulantes y antiplaquetarios, depen-diendo de si la trombosis es profunda o superficial, recomendándose la anticoagulación a largo plazo por la frecuencia de las recurrencias observadas.(14-15)
En conclusión, el SAF continúa siendo en muchos aspectos una enfermedad poco conocida, a pesar de la intensa investigación que se ha realizado en los últimos años. Si bien se cita asociado a enfermedades autoinmunes, es poco frecuente su asociación con AR, por lo cual presentamos este caso, de manera a tenerlo en cuenta en pacientes con cuadros trombóticos y enfermedades inmunológicas asociadas. En la medida que exista una mejor comprensión de la inmunopatogenia, nuevas orientaciones terapéuticas podrían desarrollarse para mejorar no solo el tratamiento si no también la prevención del desarrollo de este síndrome.
BIBLIOGRAFÍA
(1) Marini M, Rossi G, Magariños G. Síndrome antifosfolipídico: presentación de un caso y actualización del tema. Med Cutan Iber Lat Am 2002;30(5):111-5.
(2) Lashak C, Leroux MB. Anticuerpos antifosfolípidos: Valoración clínica actual. Arch. Argent. Dermatol. 2009, 59:107-113.
(3) García Rincón CI, Morales Restrepo N, Muñoz-Grajales C, Cardozo Avendaño SL, Calle J, Velazquez Franco CJ et al. Necrosis cutánea extensa secundaria a síndrome antifosfolípido en un paciente con infección por el virus de la inmunodeficiencia humana: reporte de caso. Rev Colomb Reumatol. 2014; 21(3):155-159.
(4) García-García C. Anticuerpos antifosfolípidos y síndrome antifosfolípido: actitudes diagnósticas y terapéuticas. Actas Dermosifiliogr. 2007; 98:16-23.
(5) Barraza S, Estrella V, Leroux MB et al. Síndrome anticuerpo antifosfolípido. ¿Cómo llegamos a su diagnóstico? Dermat Argent 2000; 6: 342-349.
(6) Leroux MB, Barraza S, Estrella V y Bearzotti M. Síndrome anticuerpo antifosfolípido. Arch Argent Dermatol. 2000; 50:109-115.
(7) Shoenfeld Y, Meroni P, Cervera R. Antiphospholipid syndrome dilemmas still to be solved: 2008 status. Ann RheumDis. 2008; 67: 438-442.
(8) Alfaro R. Síndrome antifosfolipídico, revisión bibliográfica. Rev Médica C osta Rica y Centroamérica. 2009; 66 (589): 313-317.
(9) O mar P, Yalili C, Iarmila C. Síndrome de anticuerpos antifosfolípidos. MEDISAN 2012; 16(3):429-444.
(10) Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for the antiphospholipid syndrome. J Rheumatol. December 2006; 33(12):2379-2382.
(11) R odríguez Santamaría J, Badziak D, Ferreirade Barros M, Luiz Mandelli F, Cavalin LC, Shigueru Sato M. Síndrome antifosfolípide. An Bras Dermatol. 2005; 80(3): 3-4.
(12) Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R. International consensus statement on an update of the classification criteria for definitive antiphospholipid syndrome. J Thromb Haemost; 4 (2): 295-306.
(13) C ontreras M. Inmunopatogenia del sindrome antifosfolipídico. Rev. chil. reumatol. 2009; 25(4):149-155.
(14) Lim W. Antiphospholipid syndrome. Hematology 2013:675-80.
(15) Cuadrado MJ, Bertolaccini ML, Seed PT, et al. Low-dose aspirin vs low-dose aspirin plus low-intensity warfarin in thromboprophylaxis: a prospective, multicentre, randomized, open, controlled trial in patients positive for antiphospholipid antibodies (ALIWAPAS). R heumatology 2014;53:275-84.
Fecha de envío: 10/05/2015 - Fecha de aprobación: 30/05/2015